OpenVax
Open source software for personalized cancer vaccines
Contributors



Publications

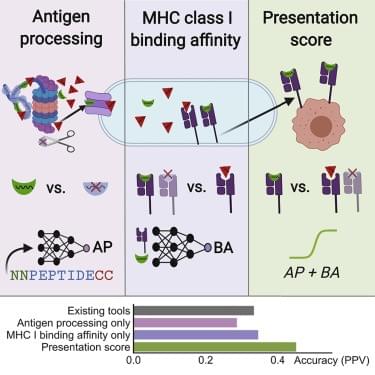
We developed an integrated predictor of MHC class I presentation that combines new models for MHC class I binding and antigen processing. Considering only peptides first predicted by the binding model to bind strongly to MHC, the antigen processing model is trained to discriminate published mass spectrometry-identified MHC class I ligands from unobserved peptides. The integrated model outperformed the two individual components as well as NetMHCpan 4.0 and MixMHCpred 2.0.2 on held-out mass spectrometry experiments. Our predictors are implemented in the open source MHCflurry package, version 2.0 (github.com/openvax/mhcflurry).

This paper describes the sequencing protocol and computational pipeline for the PGV-001 personalized vaccine trial. PGV-001 is a therapeutic peptide vaccine targeting neoantigens identified from patient tumor samples. Peptides are selected by a computational pipeline which identifies mutations from tumor/normal exome sequencing and ranks mutant sequences by a combination of predicted Class I MHC affinity and abundance estimated from tumor RNA. The PGV pipeline is modular and consists of independently usable tools and software libraries. We hope that the functionality of these tools may extend beyond the specifics of the PGV-001 trial and enable other research groups in their own neoantigen investigations.

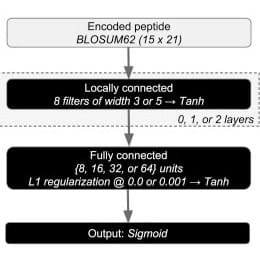
Predicting the binding affinity of major histocompatibility complex I (MHC I) proteins and their peptide ligands is important for vaccine design. We introduce an open-source package for MHC I binding prediction, MHCflurry. The software implements allele-specific neural networks that use a novel architecture and peptide encoding scheme. When trained on affinity measurements, MHCflurry outperformed the standard predictors NetMHC 4.0 and NetMHCpan 3.0 overall and particularly on non-9-mer peptides in a benchmark of ligands identified by mass spectrometry. The released predictor, MHCflurry 1.2.0, uses mass spectrometry datasets for model selection and showed competitive accuracy with standard tools, including the recently released NetMHCpan 4.0, on a small benchmark of affinity measurements. MHCflurry's prediction speed exceeded 7,000 predictions per second, 396 times faster than NetMHCpan 4.0. MHCflurry is freely available to use, retrain, or extend, includes Python library and command line interfaces, may be installed using package managers, and applies software development best practices.